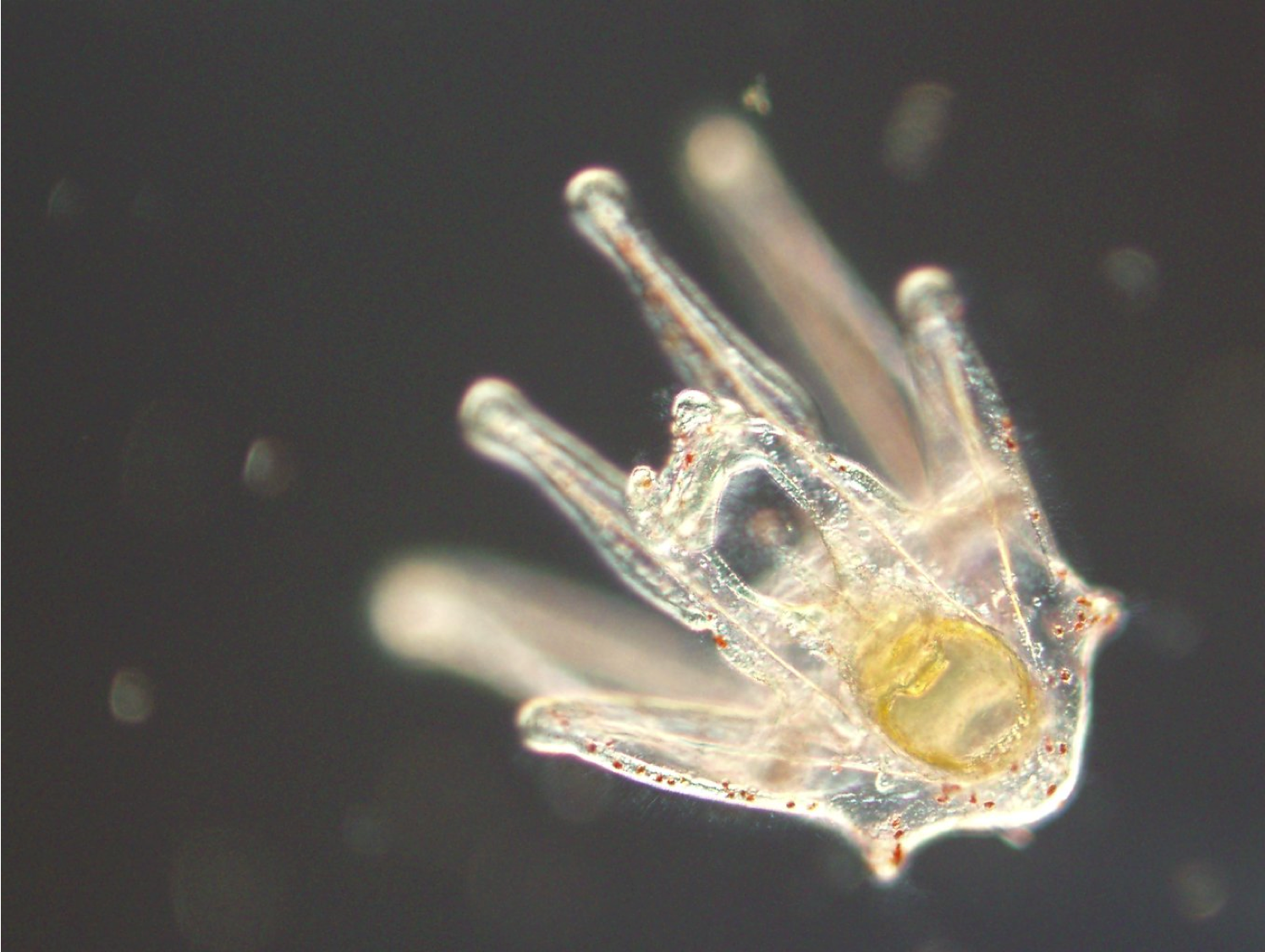
Possibilities of skin coat color-dependent risks and risk factors of squamous cell carcinoma and deafness of domestic cats inferred via RNA-seq data
The transcriptome data of skin cells from domestic cats with brown, orange, and white coats were analyzed using a public database to investigate the possible relationship between coat color-related gene expression and squamous cell carcinoma risk, as well as the mechanism of deafness in white cats. We found that the ratio of the expression level of genes suppressing squamous cell carcinoma to that of genes promoting squamous cell carcinoma might be considerably lower than the theoretical estimation in skin cells with orange and white coats in white-spotted cat. We also found the possibility of the frequent production of KIT lacking the first exon (d1KIT) in skin cells with white coats, and d1KIT production exhibited a substantial negative correlation with the expression of SOX10, which is essential for melanocyte formation and adjustment of hearing function. Additionally, the production of d1KIT was expected to be due to the insulating activity of the feline endogenous retrovirus 1 (FERV1) LTR in the first intron of KIT by its CTCF binding sequence repeat. These results contribute to basic veterinary research to understand the relationship between cat skin coat and disease risk, as well as the underlying mechanism.
(Awazu, et al., Possibilities of skin coat color-dependent risks and risk factors of squamous cell carcinoma and deafness of domestic cats inferred via RNA-seq data. Genes to Cells 28. 893-905 (2023))
Sequence and functional analysis of sea urchin CTCF --- The ancient face of a universal(?) nuclear chromosome structure regulator? ---
Mathematical model of cell state-dependent morphological changes of nuclear speckles
Nuclear speckles are nuclear bodies consisting of populations of small and irregularly shaped droplet-like molecular condensates that contain various splicing factors. Recent experiments have revealed the following structural features of nuclear speckles: (I) Each molecular condensate contains SON and SRRM2 proteins, and MALAT1 non-coding RNA surrounds these condensates; (II) During normal interphase of the cell cycle in multicellular organisms, these condensates are broadly distributed throughout the nucleus. In contrast, when cell transcription is suppressed, the condensates fuse and form strongly condensed spherical droplets; (III) SON is dispersed spatially in MALAT1 knocked-down cells and MALAT1 is dispersed in SON knocked-down cells because of the collapse of the nuclear speckles. However, the detailed interactions among the molecules that are mechanistically responsible for the structural variation remain unknown. In this study, a coarse-grained molecular dynamics model of the nuclear speckle was developed by considering the dynamics of SON, SRRM2, MALAT1, and pre-mRNA as representative components of the condensates. The simulations reproduced the structural changes, which were used to predict the interaction network among the representative components of the condensates.
( Wakao, et al., Mathematical model of structural changes in nuclear speckle. Biophys. and Physicobiol. (2023) 20, e200020 )
A mathematical model that forms a network with "modularity" and "small-world nature" similar to brain neural networks.
In this study, we performed comprehensive morphological investigations of spontaneously formed network structures among elements in coupled map systems involving global connections that change depending on the synchronicity of states of elements and spatially local connections. The model formed various hierarchical networks, some of which were classified as small-world networks containing multiple module networks, similar to the neural network of mammalian brains. Moreover, such complex networks were formed in wider parameter regions when the global connection to one element from the other element was strengthened by the synchronization between the present and past states of the former and latter elements, respectively. This study suggests that the time delay effects for connection changes among elements and local interactions promoted the self-organization of small-world networks containing module networks, such as neural networks; neural networks contain them as spike-timing-dependent plasticity and inter-neuron interaction through glial cells.
( Nakanishi, et al., Formation of Small-World Network Containing Module Networks in Globally and Locally Coupled Map System with Changes in Global Connection with Time Delay Effects. J. Phys. Soc. Jpn. 92, 034801 (2023) )
Mechanism that drives transient pairing of X chromosomes during the cell differentiation process of mouse ES cells
X chromosome inactivation center (Xic) pairing occurs during the differentiation of embryonic stem (ES) cells from female mouse embryos, and is related to X chromosome inactivation, the circadian clock, intra-nucleus architecture, and metabolism. However, the mechanisms underlying the identification and approach of X chromosome pairs in the crowded nucleus are unclear. To elucidate the driving force of Xic pairing, we developed a coarse-grained molecular dynamics model of intranuclear chromosomes in ES cells and in cells 2 days after the onset of differentiation (2-day cells) by considering intrachromosomal epigenetic-structural feature-dependent mechanics. The analysis of the experimental data showed that X-chromosomes exhibit the rearrangement of their distributions of open/closed chromatin regions on their surfaces during cell differentiation. By simulating models where the excluded volume effects of closed chromatin regions are stronger than those of open chromatin regions, such rearrangement of open/closed chromatin regions on X-chromosome surfaces promoted the mutual approach of the Xic pair. These findings suggested that local intrachromosomal epigenetic features may contribute to the regulation of cell species-dependent differences in intranuclear architecture.
( Komoto, et al., Epigenetic-structural changes in X chromosomes promote Xic pairing during early differentiation of mouse embryonic stem cells. Biophys. and Physicobiol. (2022) 19, e190018 )
Mechanisms driving gastrula invagination during sea urchin morphogenesis
Gastrulation is a universal process in the morphogenesis of many animal embryos. Although morphological and molecular events in gastrulation have been well studied, the mechanical driving forces and underlying regulatory mechanisms are not fully understood. Here, we investigated the gastrulation of embryos of a sea urchin, Hemicentrotus pulcherrimus, which involves the invagination of a single-layered vegetal plate into the blastocoel. We observed that omeprazole, a proton pump inhibitor capable of perturbing the left-right asymmetry of sea urchin embryo, induced "partial exogastrulation" where the secondary invagination proceeds outward. During early gastrulation, intracellular apical-basal polarity of F-actin distribution in vegetal half was higher than those in animal half, while omeprazole treatment disturbed the apical-basal polarity of F-actin distribution in vegetal half. Furthermore, gastrulation stopped and even partial exogastrulation did not occur when F-actin polymerization or degradation in whole embryo was partially inhibited via RhoA or YAP1 knockout. A mathematical model of the early gastrulation reproduced the shapes of both normal and exogastrulating embryos using cell-dependent cytoskeletal features based on F-actin. Additionally, such cell position-dependent intracellular F-actin distributions might be regulated by intracellular pH distributions. Therefore, apical-basal polarity of F-actin distribution disrupted by omeprazole may induce the partial exogastrulation via anomalous secondary invagination.
( Watanabe, et al., Partial exogastrulation due to apical-basal polarity of F-actin distribution disruption in sea urchin embryo by omeprazole. Geneds to Cells (2022) https://doi.org/10.1111/gtc.12934 )
Elucidation of the mechanical mechanism of dynamic genome structural reorganization during DNA double-strand break repair in yeast
During the repair of double-strand breaks (DSBs) in DNA, active mobilizations for conformational changes in chromosomes have been widely observed in eukaryotes, from yeast to animal and plant cells. DSB-damaged loci in the yeast genome showed increased mobility and relocation to the nuclear periphery. However, the driving forces behind DSB-induced chromatin dynamics remain unclear. In this study, mathematical models of normal and DSB-damaged yeast chromosomes were developed to simulate their structural dynamics. The effects of histone degradation in the whole nucleus and the change in the physical properties of damaged loci due to the binding of SUMOylated repair proteins were considered in the model of DSB-induced chromosomes based on recent experimental results. The simulation results reproduced DSB-induced changes to structural and dynamical features by which the combination of whole nuclear histone degradation and the rigid structure formation of repair protein accumulations on damaged loci were suggested to be primary contributors to the process by which damaged loci are relocated to the nuclear periphery.
( Nakahata, et al., Mathematical model of chromosomal dynamics during DNA double strand break repair in budding yeast. Biophys. and Physicobiol. (2022) 19, e190012 )
Elucidation of factors and mechanisms involved in controlling the structure of the nucleolus
The nucleolus is the site of ribosome assembly and formed through liquid-liquid phase separation. Multiple ribosomal DNA (rDNA) arrays are bundled in the nucleolus, but the underlying mechanism and significance are unknown. In the present study, we performed high-content screening followed by image profiling with the wndchrm machine learning algorithm. We revealed that cells lacking a specific 60S ribosomal protein set exhibited common nucleolar disintegration. The depletion of RPL5 (also known as uL18), the liquid-liquid phase separation facilitator, was most effective, and resulted in an enlarged and un-separated sub-nucleolar compartment. Single-molecule tracking analysis revealed less-constrained mobility of its components. rDNA arrays were also unbundled. These results were recapitulated by a coarse-grained molecular dynamics model. Transcription and processing of ribosomal RNA were repressed in these aberrant nucleoli. Consistently, the nucleoli were disordered in peripheral blood cells from a Diamond-Blackfan anemia patient harboring a heterozygous, large deletion in RPL5. Our combinatorial analyses newly define the role of RPL5 in rDNA array bundling and the biophysical properties of the nucleolus, which may contribute to the etiology of ribosomopathy.
( Matsumori, et al., Ribosomal protein L5 facilitates rDNA-bundled condensate and nucleolar assembly. Life Sci. Alli. 5, e202101045 (2022) )
Morphology of plastic chaotic coupled systems with large degrees of freedom towards understanding the self-organization of neural networks in the brain
Elucidation of the mechanism of sensitive activation regulation of transcriptional regulator (FACT) with a intrinsically disordered region
Facilitates chromatin transcription (FACT) is a histone chaperone that functions as a nucleosome remodeler and a chaperone. The two subunits of FACT, Spt16 and SSRP1, mediate multiple interactions between the subunits and components of the nucleosome. Among the interactions, the role of the DNA-binding domain in SSRP1 has not been characterized. We reported previously that the DNA-binding domain in Drosophila SSRP1 (dSSRP1) has multiple casein kinase II phosphorylation sites, and the DNA binding affinity of the domain changes sigmoidally in response to the degree of phosphorylation ("ultrasensitive response"). In this report, we explored the molecular mechanisms for the ultrasensitive response of the DNA-binding domain in dSSRP1 using the shortest fragment (AB-HMG, residues 434-624) responsible for nucleosome binding. AB-HMG contains two intrinsically disordered (ID) regions: the N-terminal part rich in acidic residues (AID) and the C-terminal part rich in basic residues (BID) followed by the HMG box. NMR and coarse-grained molecular dynamics simulations revealed a phosphorylation-dependent change in intramolecular contacts between the AID and BID-HMG, which is mediated by a hinge bending motion of AB-HMG to enable the ultrasensitive response. Ultrasensitivity generates two distinct forms of dSSRP1, which are high- and low-affinity nucleosome-binding forms. Drosophila FACT (dFACT) switches function according to the degree of phosphorylation of the AID in dSSRP1. We propose that dFACT in various phosphorylation states functions cooperatively to facilitate gene regulation in the context of the chromatin.( Aoki, et al., Ultrasensitive Change in Nucleosome Binding by Multiple Phosphorylations to the Intrinsically Disordered Region of the Histone Chaperone FACT. J. Mol. Biol. 432 (2020) 4637-4657. )
Mechanism of formation of rhodopsin linear supramolecular structure in the disc membrane of vertebrate retinal rod cells
The visual photopigment protein rhodopsin (Rh) is a typical G protein-coupled receptor (GPCR) that initiates the phototransduction cascade in retinal disk membrane of rod-photoreceptor cells. Rh molecule has a tendency to form dimer, and the dimer tends to form rows, which is suggested to heighten phototransduction efficiency in single-photon regime. In addition, the dimerization confers Rh an affinity for lipid raft, i.e. raftophilicity. However, the mechanism by which Rh-dimer raftophilicity contributes to the organization of the higher order structure remains unknown. In this study, we performed coarse-grained molecular dynamics simulations of a disk membrane model containing unsaturated lipids, saturated lipids with cholesterol, and Rh-dimers. We described the Rh-dimers by two-dimensional particle populations where the palmitoyl moieties of each Rh exhibits raftophilicity. We simulated the structuring of Rh in a disk for two types of Rh-dimer, i.e., the most and second most stable Rh dimers, which exposes the raftophilic regions at the dimerization-interface (H1/H8 dimer) and two edges away from the interface (H4/H5 dimer), respectively. Our simulations revealed that only the H1/H8 dimer could form a row structure. A small number of raftophilic lipids recruited to and intercalated in a narrow space between H1/H8 dimers stabilize the side-by-side interaction between dimers in a row. Our results implicate that the nano-sized lipid raft domains act as a "glue" to organize the long row structures of Rh-dimers.( Kaneshige, et al., Affinity of rhodopsin to raft enables the aligned oligomer formation from dimers: Coarse-grained molecular dynamics simulation of disk membranes. PLoS ONE 15 (2020) e0226123. )
Creation of white sea urchin (Albino Hemicentrotus pulcherrimus) through genome editing
(The main part of the research was carried out by Prof. Naoaki Sakamoto of the Molecular Genetics Laboratory.)
Sea urchins are used as a model organism for research on developmental biology and gene regulatory networks during early development. Gene knockdown by microinjection of morpholino antisense oligonucleotide (MASO) has been used to analyze gene function in early sea urchin embryos. However, as the effect of MASO is not long lasting, it is impossible to perturb genes expressed during late development by MASO. Recent advances in genome editing technologies have enabled gene modification in various organisms. We previously reported genome editing in the sea urchin Hemicentrotus pulcherrimus using zinc-finger nuclease (ZFN) and transcription activator-like effector nuclease (TALEN); however, the efficiencies of these technologies were not satisfactory. Here, we applied clustered regularly interspaced short palindromic repeat (CRISPR)-CRISPR-associated nuclease 9 (Cas9) technology to knock out the Pks1 gene in H. pulcherrimus. When sgRNAs targeting Pks1, which is required for the biosynthesis of larval pigment, were microinjected into fertilized eggs with SpCas9 mRNA, high-efficiency mutagenesis was achieved within 24 hr post fertilization and SpCas9/sgRNA-injected pluteus larvae had an albino phenotype. One of the sgRNAs yielded 100% mutagenesis efficiency, and no off-target effect was detected. In addition, the albino phenotype was maintained in juvenile sea urchins after metamorphosis, and the knockout sea urchins survived for at least one year and grew to albino adult sea urchins. These findings suggest that knockout adult sea urchins were successfully established and the CRISPR-Cas9 system is a feasible method for analyzing gene functions from late developmental to adult stage.( Liu, et al., Establishment of knockout adult sea urchins by using a CRISPR-Cas9 system. Development Growrh and Differentiation 10.1111/dgd.12624.)
Identification of novel functional non-coding DNA regions (NENLIS) in eukaryotic genomes
Chromosomes consist of various domains with different transcriptional activities separated by chromatin boundary sequences such as insulator sequences. Recent studies suggested that CTCF or other chromatin loop-forming protein binding sequences represented typical insulators. Alternatively, some long nucleosome-excluding DNA sequences were also reported to exhibit insulator activities in yeast and sea urchin chromosomes, although specific binding of loop-forming proteins was not expected for them. However, the mechanism of the insulator activities of these sequences and the possibilities of similar insulators existing in other organisms remained unclear. In this study, we first constructed and performed simulations of a coarse-grained chromatin model containing nucleosome-rich and nucleosome-excluding DNA regions. We found that a long nucleosome-excluding region between two nucleosome-rich regions could markedly hinder the associations of two neighboring chromatin regions owing to the stronger long-term-averaged rigidity of the nucleosome-excluding region compared to that of nucleosome-rich regions. Subsequent analysis of the genome-wide nucleosome positioning, protein binding, and DNA rigidity in human cells revealed that some nucleosome-excluding rigid DNA sequences without bound chromatin looping proteins could exhibit insulator activities, functioning as chromatin boundaries in various regions of human chromosomes.( Matsushima, et al., Insulator Activities of Nucleosome-Excluding DNA Sequences Without Bound Chromatin Looping Proteins. J. Phys. Chem. B 2019, 123, 5, 1035-1043.)
Role of nuclear horsetail movement in homologous pair formation during meiosis in fission yeast
Homologous sets of parental chromosomes must pair during meiosis to produce recombined sets of chromosomes for their progeny. This is accompanied by nuclear oscillatory movements. This study aims to elucidate the significance of these movements with a model, wherein external force is applied to the oscillating nucleus and via hydrodynamic interactions within the nucleus. Simulations revealed that a major force for aligning homologous chromosomes is the length-dependent sorting during chromosomal torsional turning, which occurs when the nucleus reverses the direction of its movement.( K. Takao, et al., Torsional turning motion of chromosomes as an accelerating force to align homologous chromosomes during meiosis. J. Phys. Soc. Jpn. (2019) 88, 023801 (Movie 1, Movie 2))
Relationship between interindividual expression fluctuations, regulation, and function of genes in higher multicellular organisms (Arabidopsis thaliana)
Gene expression levels exhibit stochastic variations among genetically identical organisms under the same environmental conditions. In many recent transcriptome analyses based on RNA sequencing (RNA-seq), variations in gene expression levels among replicates were assumed to follow a negative binomial distribution, although the physiological basis of this assumption remains unclear. In this study, RNA-seq data were obtained from Arabidopsis thaliana under eight conditions (21–27 replicates), and the characteristics of gene-dependent empirical probability density function (ePDF) profiles of gene expression levels were analyzed. For A. thaliana and Saccharomyces cerevisiae, various types of ePDF of gene expression levels were obtained that were classified as Gaussian, power law-like containing a long tail, or intermediate. These ePDF profiles were well fitted with a Gauss-power mixing distribution function derived from a simple model of a stochastic transcriptional network containing a feedback loop. The fitting function suggested that gene expression levels with long-tailed ePDFs would be strongly influenced by feedback regulation. Furthermore, the features of gene expression levels are correlated with their functions, with the levels of essential genes tending to follow a Gaussian-like ePDF while those of genes encoding nucleic acid-binding proteins and transcription factors exhibit long-tailed ePDF.( A. Awazu, et al., Broad distribution spectrum from Gaussian to power law appears in stochastic variations in RNA-seq data. Scientific Reports 8, (2018) 8339. )
Analysis of nuclear chromosome structural changes depending on the developmental stage of early sea urchin embryos
The nuclear positioning and chromatin dynamics of eukaryotic genes are closely related to the regulation of gene expression, but they have not been well examined during early development, which is accompanied by rapid cell cycle progression and dynamic changes in nuclear organization, such as nuclear size and chromatin constitution. In this study, we focused on the early development of the sea urchin Hemicentrotus pulcherrimus and performed three-dimensional fluorescence in situ hybridization of gene loci encoding early histones (one of the types of histone in sea urchin). There are two non-allelic early histone gene loci per sea urchin genome. We found that during the morula stage, when the early histone gene expression levels are at their maximum, interchromosomal interactions were often formed between the early histone gene loci on separate chromosomes and that the gene loci were directed to locate to more interior positions. Furthermore, these interactions were associated with the active transcription of the early histone genes. Thus, such dynamic interchromosomal interactions may contribute to the efficient synthesis of early histone mRNA during the morula stage of sea urchin development.( Matsushita, et al., Dynamic changes in the interchromosomal interaction of early histone gene loci during early development of sea urchin. J. Cell Sci (2017) 130, 4097-4107. )
Changes in nuclear chromosome structure associated with cell differentiation of rod cells in nocturnal mice (nocturnal animals)
We studied the role of active deformation dynamics in cell nuclei in chromatin positioning. Model chains containing two types of regions, with high (euchromatic) or low (heterochromatic) mobility, were confined in a pulsating container simulating a nucleus showing dynamic deformations. Brownian dynamic simulations show that the positioning of low mobility regions changes from sites near the periphery to the center if the affinity between these regions and the container periphery disappears. The former and latter positionings are similar to the "conventional" and "inverted" chromatin positionings in nuclei of normal differentiated cells and cells lacking Lamin-related proteins. Additionally, nuclear dynamical deformation played essential roles in "inverted" chromatin positioning.( A. Awazu, Nuclear dynamical deformation-induced hetero- and euchromatin positioning. Phys. Rev. E 92, (2015) 032709. )











 (Simulation movie)
(Simulation movie)






